Receptor Preparation Tutorial
The first step in using FRED, HYBRID, or POSIT is the creation of an OEDocking receptor, which is a collection of docking-related information connected to a protein. A typical receptor is shown in Figure 1 below.
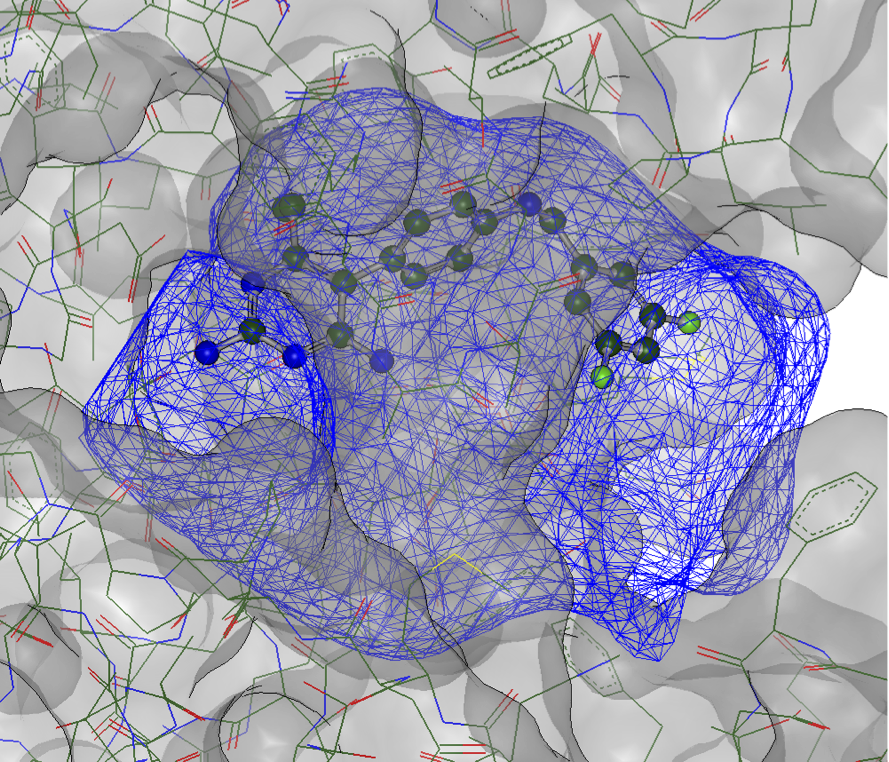
Figure 1. The OEDocking receptor and typical docking information.
There is an automated way to prepare OEDocking receptor files using SPRUCE. Alternatively, the graphical utility Make Receptor provides an interactive approach for flexible and customizable receptor preparation.
SPRUCE (Recommended)
The SPRUCE application provides functionality for automated biomolecule preparation for modeling applications. The preparation functions include tautomer evaluation, charge normalization, receptor grid generation, and missing protein loop building. A typical receptor generation session could begin with the retrieval of a set of coordinates and an experimental electron density map from the PDB (in this case, the RCSB). To automatically retrieve the data files from the PDB, the Get Structure utility in SPRUCE can be given a four character accession code.
For example, to retrieve the PDB entry 5TMG:
> getstructure 5TMG
The above command downloads 5tmg.pdb, 5tmg.cif, 5tmg.mtz files from RCSB database directly.
Once the files have been downloaded, SPRUCE can be used to generate design units (DUs) and
other receptor components. DUs are explained in the theory chapter of Spruce TK.
> spruce -in 5tmg.cif -map 5tmg.mtz
The above command creates the following intermediate DU files:
5TMG_A__DU__7EK_A-402.oedu5TMG_B__DU__7EK_B-402.oedu
DU files are generated for each of the monomers in the crystallographic asymmetric unit; in this example, there are
two copies in the asymmetric unit. If the user has supplied the electron density map (in this example, 5tmg.mtz)
to the Spruce command, the quality of the structure is evaluated using the Iridium protocols.
When there is no bound ligand detected, an error message is written into spruce_output.log.
The parameter enumsites -site_residue is required to specify a protein residue in the active site for apo
structures.
This command creates OEDocking receptor files that are readily used as docking input:
> receptorindu -in 5TMG_A__DU__7EK_A-402.oedu -out rec_5TMG.oedu
The above command then creates receptors inside the DU rec_5TMG.oedu as an input receptor file for
FRED,
HYBRID, and POSIT.
Interactive Modification of a Receptor
The Make Receptor GUI can be used to examine or modify a previously created receptor. The GUI can be used if
a user wants to define constraints or include specific water molecules in the receptor. The implementation of
Make Receptor generates an OEDesignUnit file (.oedu) for input to any of the OEDocking applications.
If using a legacy docking receptor (.oeb or .oeb.gz), conversion between the two file types
can be done using the OEDocking utility programs OEB2DUReceptor and DU2OEBReceptor. For example:
> du2oebreceptor -in rec_5TMG.oedu -out rec_5TMG.oeb.gz > oeb2dureceptor -in rec_5TMG.oeb.gz -out converted_rec_5TMG.oedu
The OEReceptor files, such as rec_5TMG.oedu, can be visualized using VIDA. The components are shown in Figure 2.
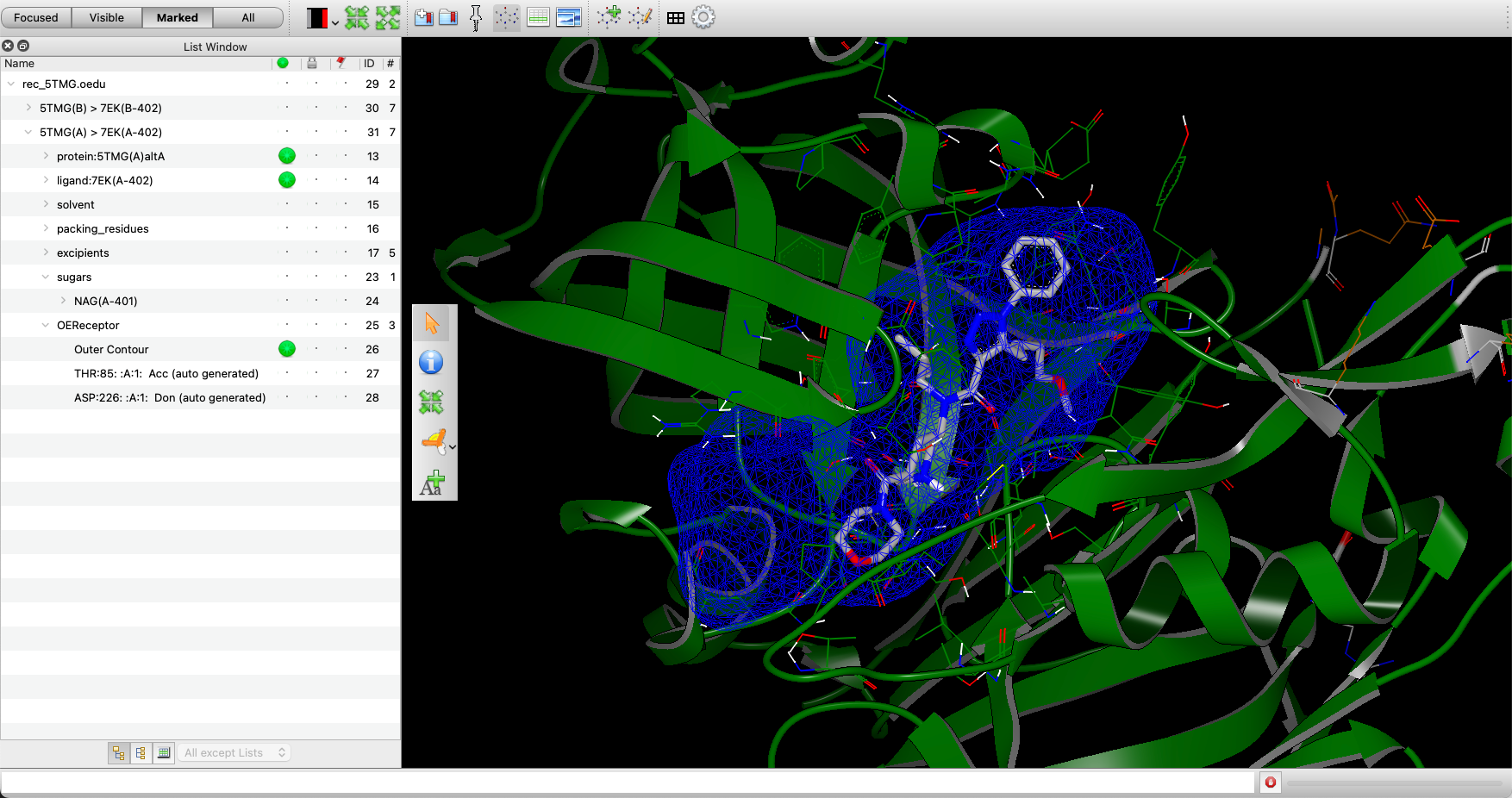
Figure 2. The view of the OEDocking receptor and components in VIDA.
This OEDocking receptor includes the protein structure and a description of the binding site. This description includes an outer contour (highlighted in the List Window), which limits the placement of heavy atoms during the FRED, HYBRID, or POSIT search procedures.
See the Make Receptor section for generating receptors from structure coordinates instead of from previously generated OEDocking receptor files.