FRED tutorial
FRED requires a protein to be docked into, a definition of a region in that protein in which the docking will take place and a multi-conformer database of molecules to be docked. The most common format for database file(s) is a multi-conformer OEBinary file created by OpenEye’s OMEGA application, however, this file can be one of several 3D formats. These formats include SDF, MOL2 and PDB. FRED determines the database file format from the file extension, .sdf or .mol for SDF, .mol2 for MOL2, .pdb or .ent for PDB. Gzip compressed files of these same formats are allowed as well. FRED will interpret infile.sdf.gz as a gzip’ed SDF file.
Note
Even though all these formats are supported, using SDF, PDB or MOL2 can result in a loss of speed due to the I/O penalty of these formats.
FRED requires a single receptor to dock ligands into. Receptors may be created with the following programs: Make Receptor, ReceptorInDU, OEB2DUReceptor, and Receptor preparation tutorial .
Note
Receptors are encapsulated inside a OEDesignUnit in .oedu files.
To dock a set of ligands into the receptor 2IKO_rec.oedu:
> fred -receptor renin/receptors/2IKO_rec.oedu -dbase renin/all.oeb.gz
By default, FRED generates several output files. Unless you specify a different
Output prefix during setup, FRED will use the prefix fred for all output files.
fred_docked.oeb.gz- top 500 scoring molecules ofall.oeb.gzdocked into2IKO_rec.oedu.fred_undocked.oeb.gz- molecules ofall.oeb.gzthat could not be docked into the active site (generally occurs if the molecules are too big for the site). This file will not be present if all molecules were successfully docked to the active site.fred_score.txt- a tab separated text file containing the name and score of each of the top 500 ligands.fred_report.txt- a text report of the docking process.fred_settings.param- a text file containing the parameters used for this run.fred_status.txt- a text file that is written periodically during the run with the status of the run.
If you have a multi-processor machine, you can harness the extra computing power by using OpenMPI. To re-run the job on a host with 4 processors, you enter the following:
> fred -mpi_np 4 -receptor renin/receptors/2IKO_rec.oedu -dbase renin/all.oeb.gz -prefix 2IKO_fred
The output from the docking runs may be viewed in VIDA, as depicted in figure 2IKO FRED Docked.
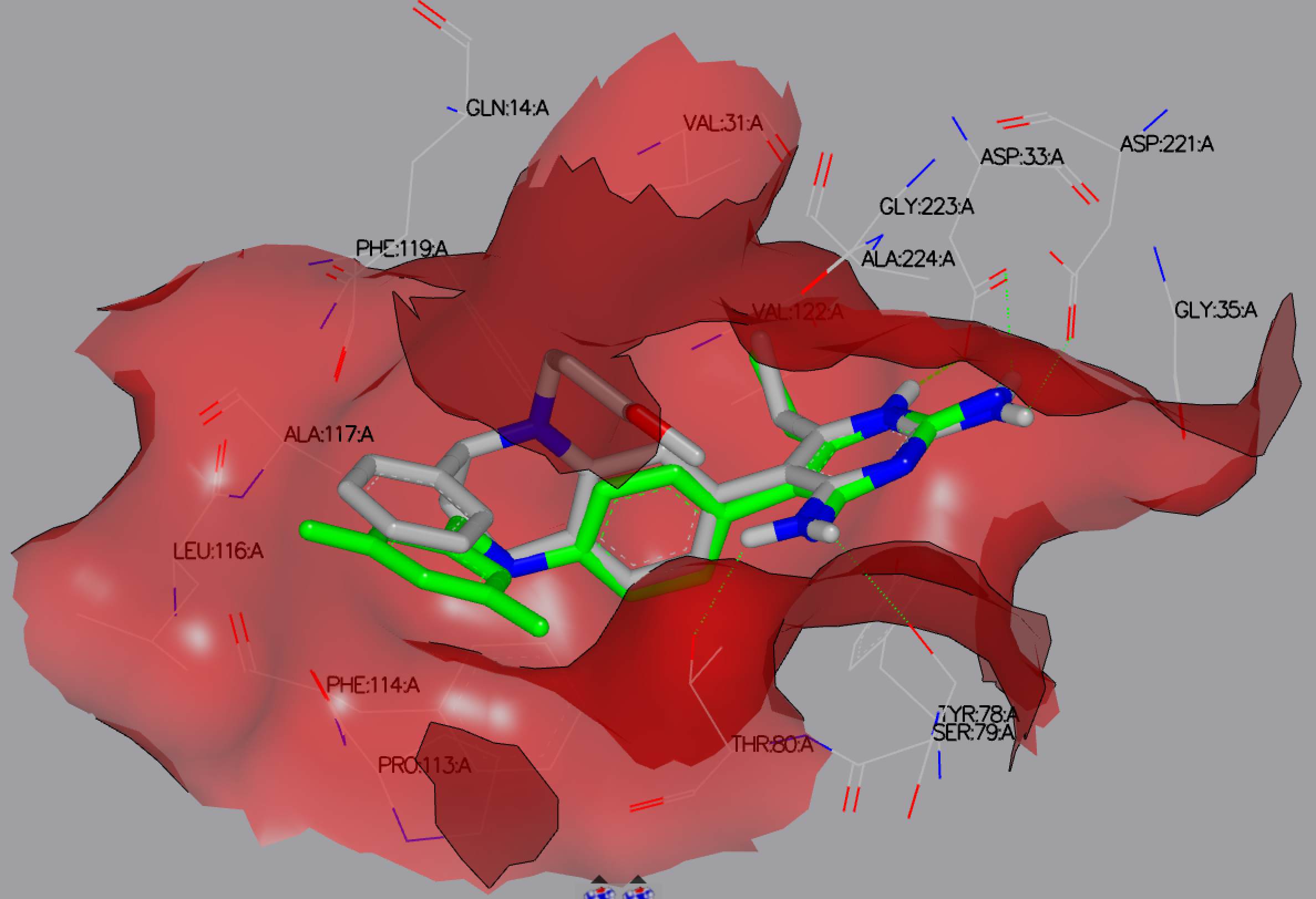
2IKO FRED Docked results showing the reference ligand (green) and the top docked ligand
In addition to analyzing and visualizing the output from a docking run in VIDA, a summary PDF report can be generated using DockingReport.
> docking_report -docked_poses 2IKO_fred_docked.oeb.gz \ -receptor renin/receptors/2IKO_rec.oedu -report_file 2IKO_fred_docked_report.pdf
The result of running the above command is depicted in figure 2IKO FRED Docking Report.
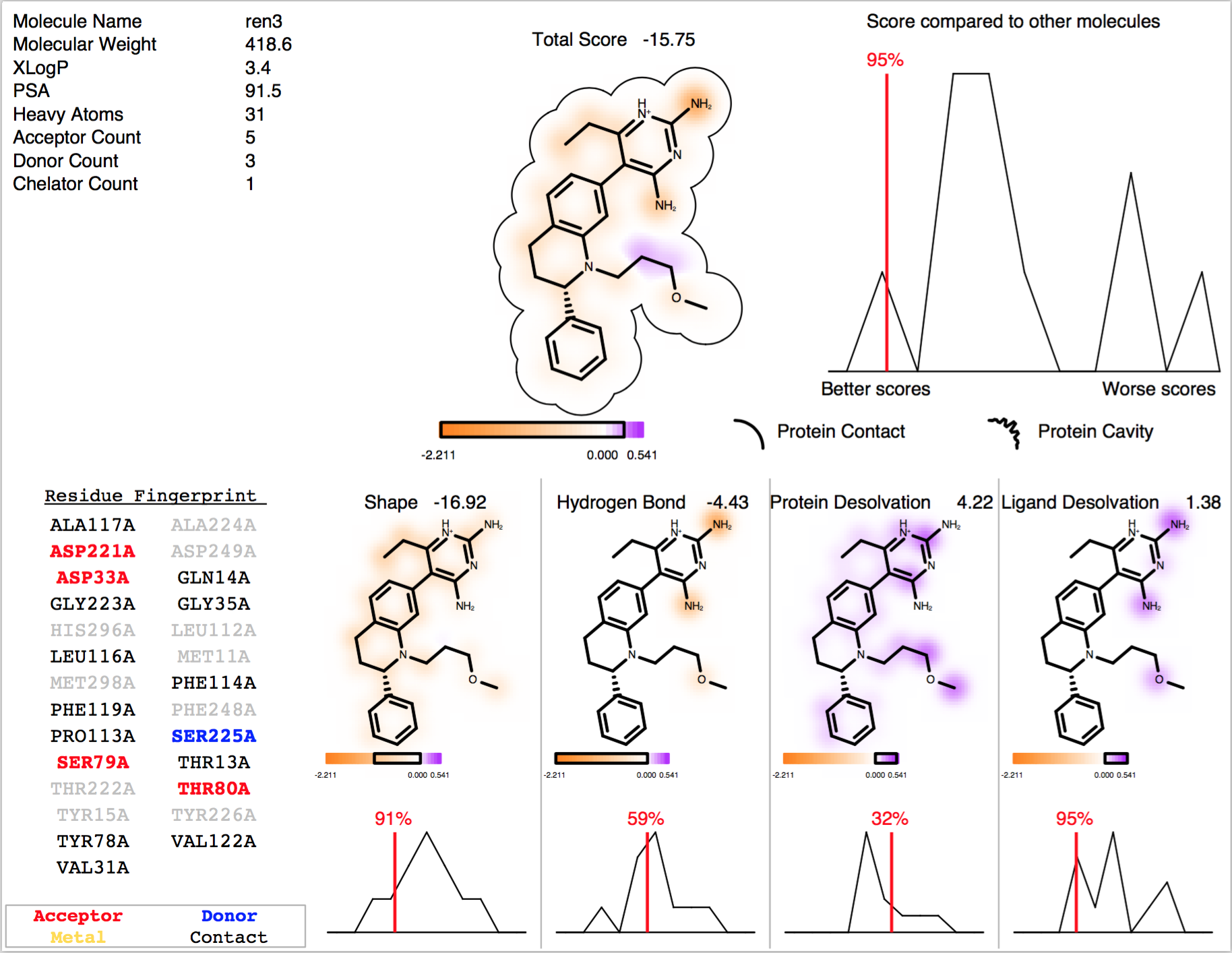
2IKO FRED Docking Report
The PDF report includes a 2D depiction of each molecule, a breakdown of the docking score components by atom, a comparison of the molecule’s score compared to the other docked molecules, and a ‘Residue Fingerprint’ which highlights which residues in the receptor site the ligand is interacting with. Greyed out residues are residues in the site that other ligands interact with, but the current ligand does not.