OEApplications 2024.1¶
Release Highlights 2024.1.0¶

EON: Overlay Optimization with Shape and Charge Density¶
EON, the tool for molecular similarity based on shape and electrostatics, has been reimagined with the addition of charge density as a descriptor for electrostatic properties. By replacing electrostatic potential with molecular charge density as the primary descriptor of electrostatic properties, EON no longer requires the input molecules to be pre-aligned with the query molecule. Instead, EON now aligns the input molecules using shape- and electrostatics-based overlay optimization, where electrostatics are modeled via molecular charge density. EON still has the option to use electrostatic potential as a measure of similarity, if desired. Figure 1 shows an example of EON overlay with charge density visualization.
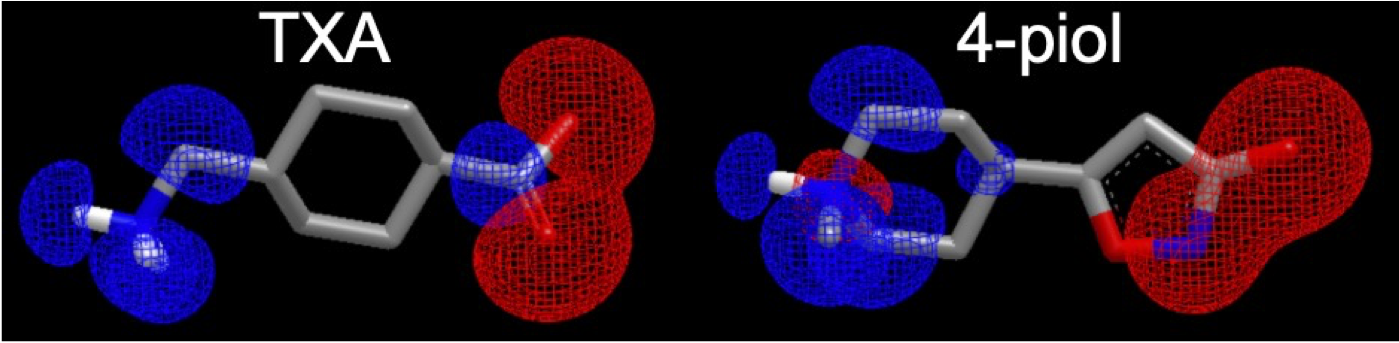
Figure 1. EON shape charge density overlay between two molecules.¶
Even with the addition of overlay optimization within the workflow, the new EON is now significantly faster compared to the previous version; and yet, as a tool for virtual screening with shape and charge similarity, the behavior of EON remains the same. When compared to ROCS, EON lags slightly when measured in terms of virtual screening success rates but excels in finding hits that are more chemically diverse.
Performance of the previous and current versions of EON, as compared to ROCS,
are displayed in Figure 2. The comparisons were performed using multi-query directory of
useful decoys (MDUD) datasets containing 38 targets. The current version of EON
overlays and scores with shape and charge similarity. A command line flag -potential supports the rescoring
of shape- and charge-aligned conformers with shape and electrostatic potential similarity. The current default version
of EON is now almost as fast as ROCS, as can be seen in the right-hand plot below.
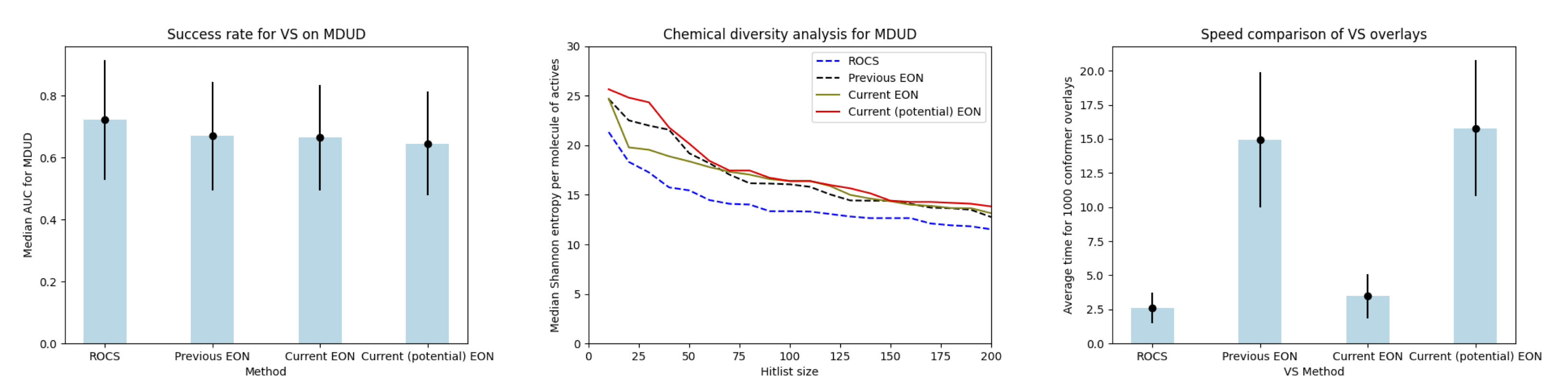
Figure 2. Comparison of performance of EON and ROCS for virtual screening success rate (left); for chemical diversity of actives found (middle); and for the speed of calculations (right). “Current” refers to the default version using shape and charge density for similarity, and “current (potential)” refers to EON using shape and electrostatics.¶
Bioisostere TK: New Toolkit for Bioisosteric Fragment Replacement¶
The 2024.1 release introduces Bioisostere TK, a new toolkit for bioisosteric analog generation. It involves replacing a portion of a lead compound with fragments that have similar shape and electrostatics but with potentially novel connectivity and chemistry. This provides toolkit level access to the existing BROOD application functionality.
Bioisostere TK also brings several additional functionalities. At its core are tools for fragment-based modeling, such as fragment creation, fragment conformer generation, and fragment alignment and similarity in 3-dimensional space. Bioisostere TK brings OpenEye’s shape-, color-, and electrostatics-based molecular similarity tools into fragment-based similarity. It also exposes tools for the generation of 3D molecule conformers with fragment replacement.
An example of fragment alignment with Bioisostere TK based on shape and color is shown in Figure 3. The attachment points in both the reference and fit fragments are marked in purple. The fragment overlay highlights how special attention is paid to the overlay of the attachment points.
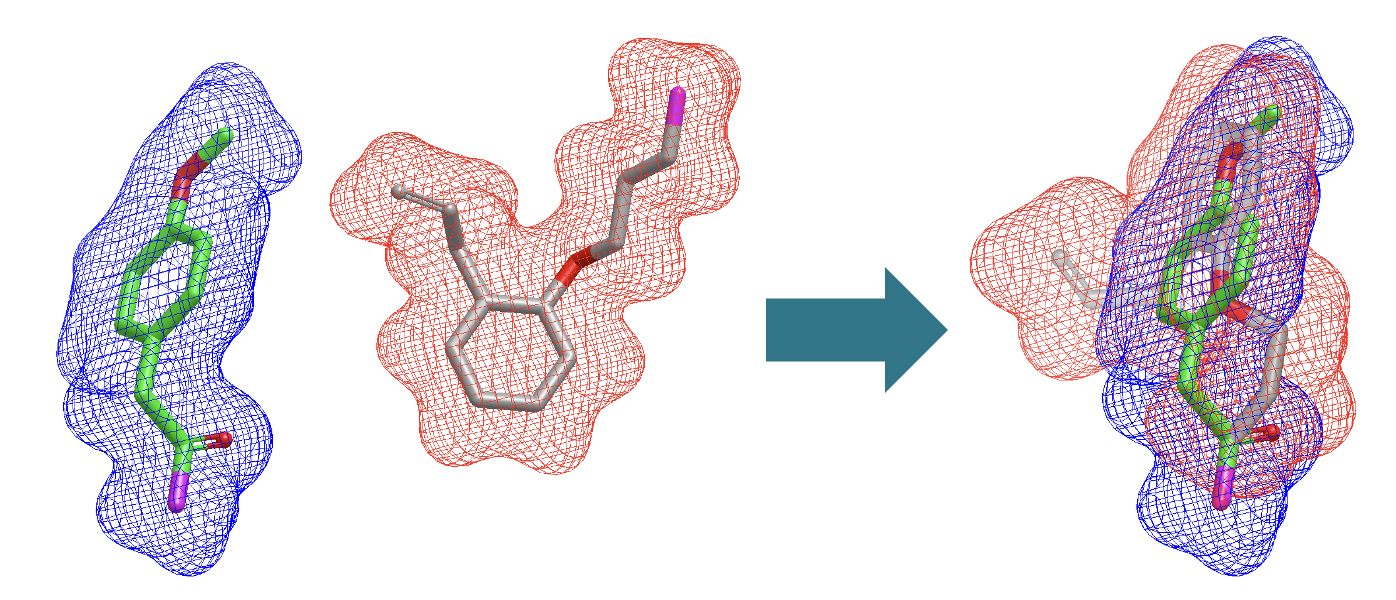
Figure 3. Fragment overlay between a reference and a fit fragment.¶
FLYNN: Ligand fitting for Cryo-EM¶
FLYNN, the tool for fitting small molecules to electron density, has been extended to support density maps generated through cryo-electron microscopy (cryo-EM).
FLYNN is a powerful tool for ligand fitting that uses adiabatic fitting to generate the best-fitting, lowest strain conformers consistent with experimental electron densities and structural models. This tool was one of the first to enable high-quality, physics-informed pockets and poses from X-ray crystallographic models. FLYNN has now been updated to enable researchers to access the same powerful insights from maps and models generated through cryo-electron microscopy (cryo-EM).
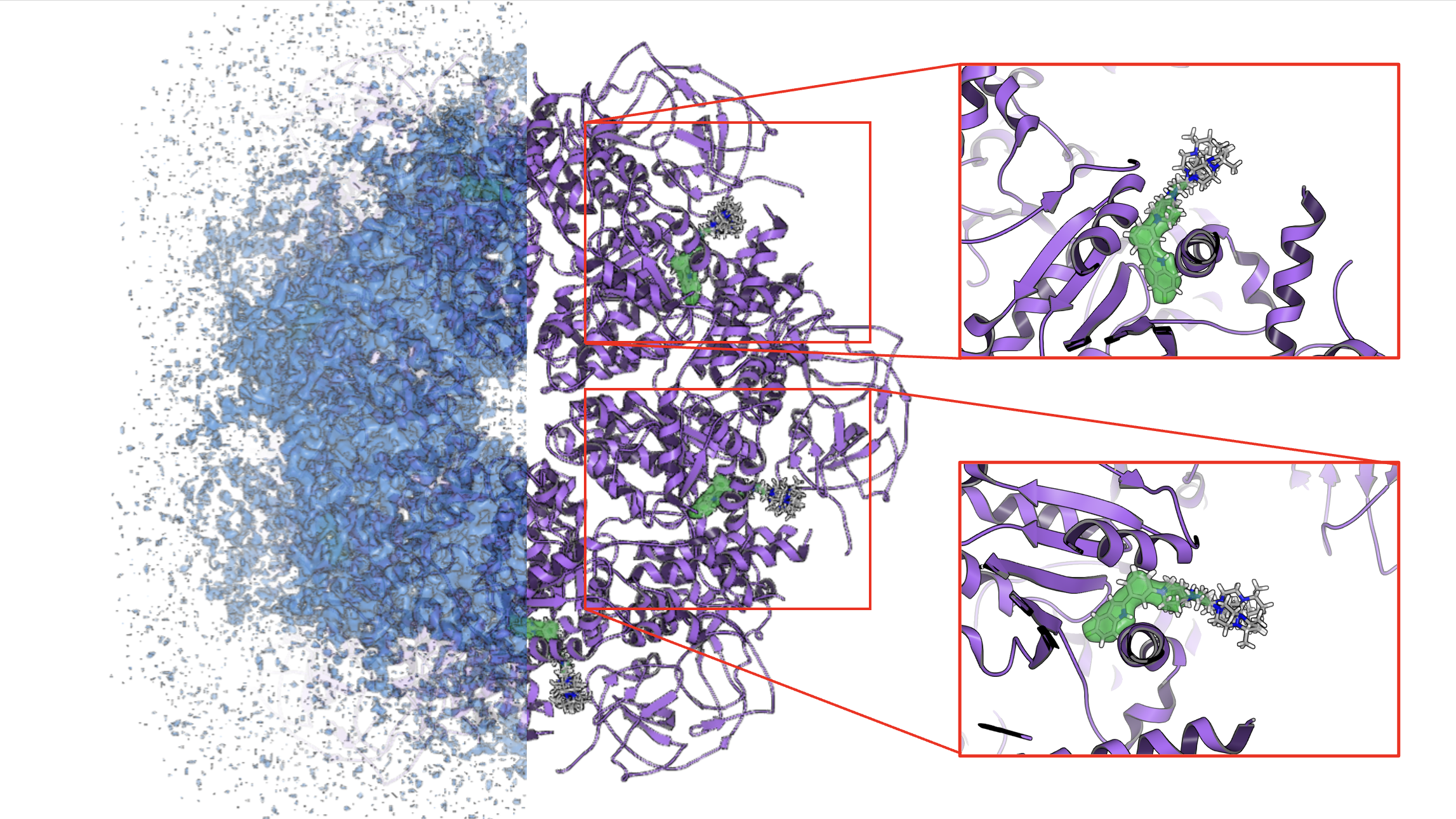
Figure 4. Ligand fitting with FLYNN to predict binding pockets and poses consistent with electron density from cryo-electron microscopy (cryo-EM).¶
Hydrogen Placement Updates in SPRUCE¶
In the 2024.1 release of SPRUCE, several improvements have been made to OEPlaceHydrogens to handle metal cation chelation effects on the nearby protonation states, specifically for ligands. The changes shown in Figure 5 below highlight new state and optimized geometries for sulfonamide, hydroxamate, phosphate, and borate groups. In addition, changes have been made around geometries for sp3 secondary amines. Lastly, improvements have been added in OEProtonateDesignUnit to allow for protonation state changes related to the detection of unfavorable acceptor–acceptor clashes, resulting in a more favorable hydrogen bond network.
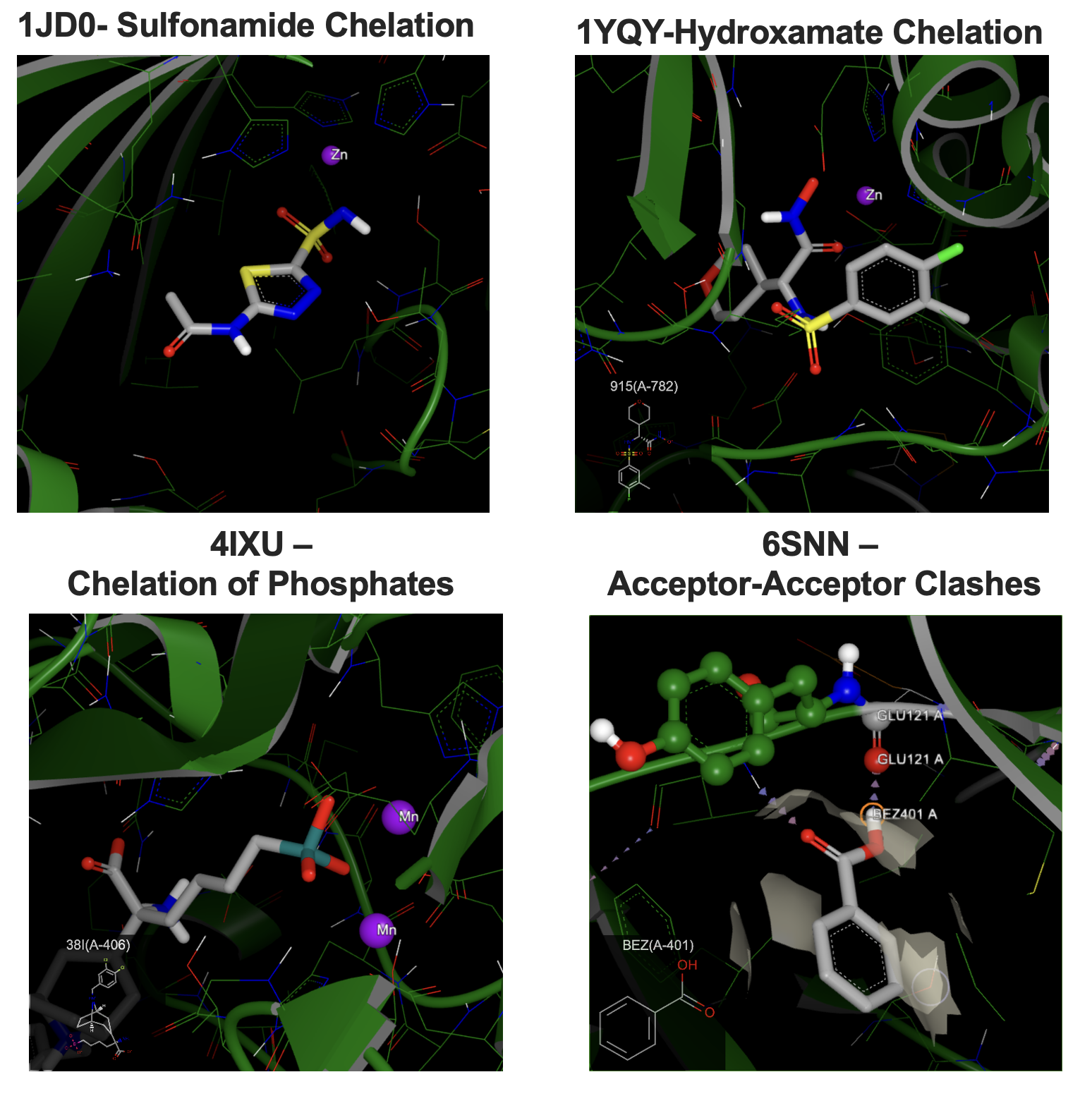
Figure 5. Examples of improved chelator interactions and deprotonation rules for different moieties: hydroxamate, sulfonamide, and phosphates/borates. Also, on the bottom right, an example of improved interaction networks with acceptor–acceptor heuristic fixes.¶
Supported Platforms¶
OS
Versions
Linux
RHEL8/9, Ubuntu20/22
Windows
Win10, Win11
macOS
12, 13, 14
Detailed Release Notes 2024.1¶
AFITT 3.0.0¶
New Features¶
FLYNN can now handle maps from cryo-electron microscopy (cryo-EM) natively. Maps from cryo-EM in the .mrc, .ccp4, or .map file formats can be input using the -map map command-line parameter, alongside the protein structure file (-prot) docked/refined into the map and the 2D or 3D ligand information for the molecule to be modeled (-in).
BROOD 4.0.0¶
Major bug fixes¶
The
Filteringbutton was removed from the vBROOD home page to prevent irregular crashes and inconsistent UI behavior.
Bioisostere TK 4.0.0¶
This is the first commercial release of Bioisostere TK.
New features¶
The following classes are available as preliminary API with the first release of this toolkit.
The following functions are available as preliminary API with the first release of this toolkit.
EON 3.0.0¶
New Features¶
EON no longer requires pre-aligned molecules. The alignment and scoring are done internally with molecular shape and charge density similarity. Optionally, via modifying the boolean command line flag potential to true, rescoring of those alignments with shape and potential similarity is performed.
Zap TK 2.4.7¶
Minor internal improvements have been made.
OEDocking 4.3.1¶
Minor internal improvements have been made.
OEDocking TK 4.3.1¶
An issue has been fixed that would occasionally cause segmentation faults when OEPosit would internally fail to generate conformers.
New methods, GetPoseRelaxOptions and SetPoseRelaxOptions, have been added to OEPositOptions to allow more flexibility in relaxing the OEPosit generated poses.
Internal algorithms have been improved in OEShapeFit to improve strain in generated poses.
OMEGA 5.1.0¶
New Features¶
MakeFragLib now generates outputs that are consistent with outputs generated from OMEGA. The following outputs are generated by default.
makefraglib_frags.oeb.gz: Output fragments library
makefraglib_log.txt: Output run log
makefraglib_param.txt: Parameter file for rerunning the job
These new command line parameters have been introduced in MakeFragLib.
-ewindowRing: Energy window for ring fragment conformer generation-rmsRing: RMS threshold for ring fragment conformer generation-startfactRing: Multiplier for number of starts during ring fragment conformer generation-timeLimit: Maximum time in seconds allowed for non-ring fragment generation-fragGen: Maximum number of conformers to generate for non-ring fragments-fragKeep: Maximum number of fragments to keep for non-ring fragments-timeLimitRing: Maximum time in seconds allowed for ring fragment generation-fragGenRing: Maximum number of conformers to generate for ring fragments-fragKeepRing: Maximum number of fragments to keep for ring fragments
Major bug fixes¶
Running OMEGA with the
-useGPU falseoption on a GPU-configured machine previously caused a memory leak. This issue has now been resolved.MakeFragLib now generates multiple conformers of fragments as expected.
OMEGA can now properly handle input
CIFfiles.
Omega TK 5.1.0¶
New features¶
A new Flipper option has been added to enumerate internally perceived relative stereocenters (e.g., 1,4-disubstituted cyclohexanes). See SetEnumRelativeAtomStereo.
Minor bug fixes¶
Stereochemistry of input molecules is now respected more consistently in OEMacrocycleBuilder.
For SetMacroCycOptions in OEFragBuilderOptions, defaults for SetFragGen, SetFragKeep, and SetStartFactor have been changed to more appropriate values of 50000, 1024, and 20, respectively.
An issue with the order of loading rules to an existing torsion library with AddTorsionLibrary has been fixed.
A duplicate entry from the
GUBAtorsion library has been removed.
MolProp TK 2.6.4¶
Minor internal improvements have been made.
PICTO 5.1.1¶
Minor internal improvements have been made.
OEDepict TK 2.5.4¶
Minor internal improvements have been made.
pKa-Prospector 1.2.3¶
Minor internal improvements have been made.
QUACPAC 2.2.4¶
Minor internal improvements have been made.
Quacpac TK 2.2.3¶
Minor internal improvements have been made.
ROCS 3.6.2¶
New Features¶
Command line flags related to the EON input generation category have been removed. This is a consequence of new behavior of EON which no longer requires pre-alignment of input molecules. In particular,
-eon_input,-eon_maxconfs,-eon_input_size, and-eon_input_fileare no longer command line flags of this version of the ROCS Application.Double-clicking a shape query .sq file or selecting
Open with vROCSwill now start vROCS with the query loaded and available for editing or use in a run.
Major bug fixes¶
Color force field files
ImplicitMillsDean.cffandExplicitMillsDean.cffshipped withApplications dataare now consistent with the internal implementation of the corresponding force fields. This update specifically fixes an issue with the definition oftertiaryAmine.2D hitlist depictions during a run in vROCS have been hidden on macOS, due to an incorrect graphical display.
Minor bug fixes¶
During query editing in vROCS, the behavior of right-clicking on an atom and
Create a Color Atomhas more appropriate restrictions and no longer causes a crash.
Shape TK 3.6.2¶
New features¶
The following preliminary API classes have been made permanent.
New methods, SetAtomStrength and GetAtomStrength, have been added that allow multiplying the contribution of shape overlap for any atoms, without adding an additional atom or Gaussian.
SiteHopper 2.1.0¶
Minor internal improvements have been made.
SiteHopper TK 2.1.0¶
New features¶
GPU-based SiteHopper is no longer used to prescreen but is enabled as a stand-alone search method.
SPRUCE 1.6.0¶
Minor internal improvements have been made.
Spruce TK 1.6.0¶
New features¶
The chemical components dictionary has been updated with latest structures from the wwPDB.
A feature has been added to OEProtonateDesignUnit to detect acceptor–acceptor clashes, modify the protonation state of one of the moieties, and reoptimize the hydrogen bond network.
The following preliminary API classes have been made permanent.
The following preliminary API functions have been made permanent.
Major bug fixes¶
A crash in OESpruceFilter has been fixed in relation to atoms that were cleaned up during the standardization process.
Minor bug fixes¶
An issue has been fixed that ensures radii are assigned to all atoms in a design unit.
Documentation changes¶
An example has been added showing how to convert FASTA files to structure metadata for structures missing the SEQRES information in the PDB metadata and MMCIF equivalent.
SZYBKI 2.7.1¶
Minor internal improvements have been made.
Szybki TK 2.7.1¶
OEFixedProteinLigandOptimizer and OEFlexProteinLigandOptimizer now propagate error better when optimization fails.
VIDA 5.0.6¶
Minor internal improvements have been made.
Previous Release Highlights¶
- Release Highlights 2023.2.3
- Release Highlights 2023.1.1
- Release Highlights 2023.1.0
- Release Highlights 2022.2.2
- Release Highlights 2022.2.1
- Release Highlights 2022.1
- Release Highlights 2021.2
- Release Highlights 2021.1
- Release Highlights 2020.2.2
- Release Highlights 2020.2
- Release Highlights 2020.1.1
- Release Highlights 2020.1
- Release Highlights 2020.0
- Release Highlights 2019.Nov
- Release Highlights 2019.May
- OEApplications 2018.Nov